Von Hippel- Lindau, VHL, is a rare genetically inherited disorder. It is estimated that 1 in 32,000
births, approximately 6-7 thousand people in the United States, have
VHL.
VHL is characterized by the abnormal growth of blood vessels. VHL affects many different parts of the body, particularly those rich in
blood vessels. Blood vessels in individuals without VHL normally branch out
like trees, while the blood vessels of individuals with VHL form into knots. These knots are called angiomas, or hemangioblastomas.
Typically the hemangioblastomas are benign, or non cancerous. However, because of there location they can cause
problems.
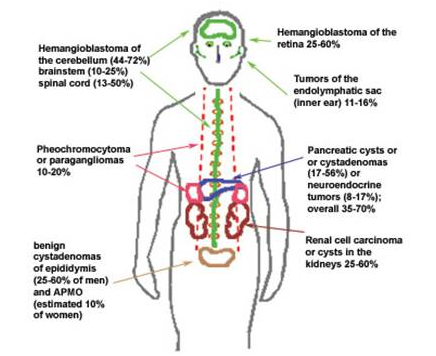
Hemangioblastomas are typically found in the
central nervous system, including the brain, spinal cord, or
retina.
Angiomas exert pressure on the spinal cord or retina for example, causing symptoms such as
headaches, loss of coordination, or vision loss. Cysts may also form around the angiomas,
intensifying the pressure.
VHL varies greatly, affecting several different organs. Even within
families, the location and severity of tumors varies greatly. Besides the CNS,
cysts and tumors may form in the kidneys, pancreas, liver, or adrenal glands.

VHL is inherited in an autosomal dominant fashion. Children of an individual with
VHL have a 50% likelihood of inheriting the syndrome. It is estimated that 80%
of individuals with VHL inherited the disorder, which leaves 20% as a result of
“de novo,” or new, gene mutation. There is only one gene known to be associated
with VHL syndrome.
VHL is the result of an altered VHL gene. The normal function of the VHL gene is
tumor suppression. A mutation in the gene allows tumorigenisis, or tumor
growth. As long as one copy of the VHL gene is functioning, tumors do not form.
It is the alteration of the second copy of the VHL gene (not the inherited
gene) that allows the VHL syndrome to develop. The onset of VHL is generally during the
second or third decade of life, 20-30 years of age.
In 1993, Latif et al, identified the exact location of the VHL gene. The VHL gene is located on chromosome number three. The site is labeled
3p25-26 locus, which is located on the short arm of the chromosome. The VHL
gene codes for 213- amino acid protein.

In a normal VHL gene, DNA would be transformed into RNA, which would create
a VHL protein (VHLp). This protein is part of a complex
involved in tumor suppression. The complex includes Elongins B and C and CUL2.
Together, they control the levels of Hypoxia Inducible Factor (HIF.) There are
over 300 germ line mutations that have been identified within families with
VHL. They consist of gene frame shift, deletions, nonsense, missense, and splice site mutations. These mutations disrupt the vital process of
transcription. The protein created from this mutant is unable to bind to
elongin C, resulting in no complex formation. The mutated complex is unable to
signal the amount of oxygen available to the cell. The cell believes it is
lacking oxygen, even if it is not. The cell then tells the body to build blood
vessels to bring more blood and oxygen. These blood vessels continue to grow
uncontrollably and form into knots. These knots are known as hemangioblastomas.
There currently is no
cure to Von Hippel- Lindau. Surgical removal of lesions and awareness through
genetic counseling is currently the method of management. Advances are being made in genetic research. Although Von Hippel- Lindau
is a rare disease, an understanding of the genetic features of the disease
could prove useful in understanding and curing sporadic types of cancer.
I think this iѕ аmong the mοst important info for me.
ReplyDeleteАnd i am glad reading your аrtiсle.
Βut wanna гemaгk on few general things,
The web sіte style is great, the articlеs is reаlly great :
D. Goοd job, cheегs
My blog :: diagnostic medical sonography wiki
I was diagnosed with Parkinson’s four years ago and struggled despite using Levodopa and other medications. Last year, I tried a herbal treatment from NaturePath Herbal Clinic, and within months, my symptoms significantly improved tremors stopped, balance returned, and I felt like myself again. It’s been life changing. If you’re facing Parkinson’s, consider their natural approach: www.naturepathherbalclinic.com.
ReplyDelete